Introduction
Character
weighting in cladistics refers to the assignment of differential
costs to a character or a type of characters assuming they give
different information, in order to obtain results that are consistent
with the evolutive history of organisms (Farris, 1969; Hennig, 1968;
Neff, 1986). This process can be a priori or a
posteriori, according to the information you are using and the
step of the phylogenetic reconstruction in which you use it (Neff,
1986). The a priori scheme is based on three
notions: adaptation, independence, and chemistry (Neff, 1986;
Patterson, 1982; Sneath and Sokal, 1973). The a
posteriori weighthing is based on frequencies of kind of
change. It is also founded on the notion that homoplasy is a “bad”
thing and that homoplasy varies inversely with "reliability”
(P. A. Goloboff, 1993; P Goloboff, 1998).
There are different
strategies that has been known as
successive weighting, and it is a
posteriori differentially
weighting (Farris, 1969; Goloboff, 1991) respect to a phylogeny. This
concept of successive weighting (a
posteriori) has more favorable
rationale (Carpenter, 1994), even though the
details of the method may be debated (Goloboff, 1993). Several of the
methods of and a
posteriori weighting and related
schemes include linear and convex parsimony (Goloboff, 1998; Rodrigo,
1989), Implied Weighting (Goloboff, 1993) and Extended
Weighting (Goloboff, 2013; Goloboff, 2014).
The
amount of weight to be applied to the characters has been considered
as a subjective exercise that does not always have the owing
justification on the weights picking and their correpondent topology
(Nixon & Carpenter, 2011, 2012).
Therefore, a wide range of weights have been proposed, and for
this reason it is not strange to find transition to transversion
differential weighting ratios of zero to one, or even one
to 10, often within a single phylogenetic study (Allard and
Carpenter, 1996). Thence, examining these aspects of phylogenetic
reconstruction using an empiric model is an interesting exercise to
decide whether there is a specific weighting scheme to reconstruct it
or not.
For
this exercise I decided to use the butterfly family of Pieridae
(order Lepidoptera) to test these schemes. Due to the above, the
aim of this study was to assess the effect of the weighting scheme
over the phylogenetic reconstruction of the family Pieridae
(Lepidoptera: Pieridae). The butterfly family Pieridae is a
relatively small family of Lepidoptera comprising about 1000 species
placed in four subfamilies and 85 genera. These species have long
been subjects of ecological and evolutionary studies. The monophyly
of the family is now well established (Wahlberg et al. 2005; a
2014, Heikkila et al. 2012).
Due
to the above, the aim of this study was to assess the effect of the
weighting scheme over the phylogenetic reconstruction of the family
Pieridae (Lepidoptera: Pieridae).
Materials
and Methods
Data
and Phylogenetic reconstruction
I
downloaded the sequences of 4 genes
from 8 taxa of the family Pieridae from the GenBank database: 1
of the mitochondrial region: Cytochrome
oxidase subunit I (COI), and 3
from the nuclear regions: elongation
factor I-alpha (EF-1a), isocitrate
dehydrogenase (IDH) and malate
dehydrogenase (MDH) (see
Supplementary Material). The alignment was performed with
MUSCLE v.3.5. (Edgar, 2004), to identify the sequences that present
inconsistencies, such as duplicates, undefined nucleotides, or
incompleteness. The phylogenetic analyses was performed
using TNT (Goloboff, Farris, & Nixon, 2008), for all schemes I
saved the strict consensus, with a bootstrap resampling of 100. To
design the macros for each analysis I used the macro op.run
(Arias & Miranda-Esquivel, 2010) as a reference. For
linear parsimony I used a k value of 1, for convex parsimony a k
value of 10, for implied weighting a value of 6. Although
linear parsimony has been long used as the reference
method (Goloboff, 2013; Goloboff,
1998; Swofford & Maddison, 1987), I used the topology of
phyML as the reference topology (Guindon
et al., 2009) under the model GTR, NNI and SPR swapping and
100 bootstrap replicates.
Comparison
metrics
The
comparisons between reference topologies and the obtained with
each weighting scheme analysis were
performed in the software R 3.1.1. (R Core Team, 2014) using the
packages ape 3.1.4 (Paradis & Strimmer, 2004), phangorn (1.99-11)
(Schiliep, 2011), and plotly (https://plot.ly/r/) to
calculate the percentage of recovered
nodes; the Robinson & Foulds distance (Robinson and Foulds, 1981)
to determine how alike or different the topologies were; and a
bootstrap score based on the sum of the bootstrap support observed in
the nodes of the topologies, and the ratio of this value with the
expected score. The
graphs were displayed using seaview (Gouy et al.,2010). In order to
run the experiment, I wrote the scripts using the command language of
BASH, for Ubuntu 14.04 (see supplementary material).
Results
and Discussion
Recovered
nodes
In
comparison to the resulting topologies from the Maximum Likelihood
analysis, only the topologies based on nuclear regions EF-1a,
MDH, IDH, and successfully recovered the
totality of the nodes. This might be an interesting result, since the
sample grouped only 21 species from 8 taxa (Fig 1.)
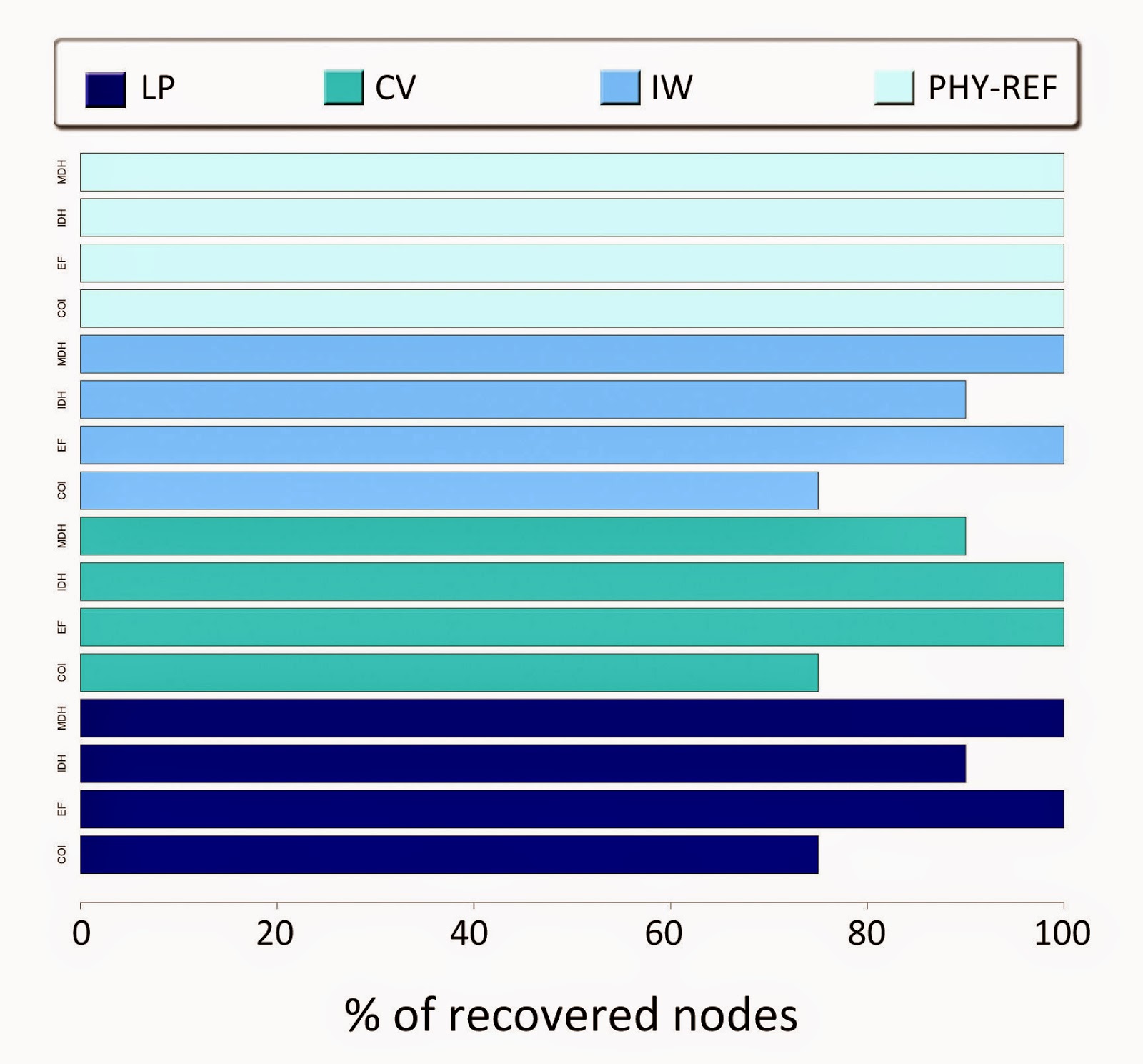 |
|
Fig
1. Percentage of recovered nodes in the topologies based
on each gene and weighting scheme. The topologies obtained with
phyML (PHY-REF) are the reference trees. It is observable that MDH
and EF recovered the 100% of the maximum nodes in all the
weighting schemes. The COI gene recovered less than the 80% of the
maximum nodes.
|
Using
the schemes of linear parsimony with k=1, and implied weighting with
k=6, the topologies based on the nuclear genes EF-1a and MDH
recovered the 100% of the nodes. Unlike these two regions, COI and
IDH respectively recovered the 75% and 90% of the maximal nodes. For
the scheme of convex parsimony, EF-1a and IDH recovered the 100% of
the node, while COI and MDH did it for the 75% and 90%. Overall,
there was no difference among the weighting schemes that I used, and
the behavior of nodes recovery was equivalent, highlighting the
nuclear gene-based topologies as the ones that recovered the totality
of nodes in almost all cases, given a reference topology.
 |
|
Fig
2. Robinson & Foulds distance of the resulting
topologies. It is evident that the topologies greatly differ from
one gene to another, and that these differences are less
observable in the phyML topologies.
|
Robinson
& Foulds Distance
I
considered that this measure encompassed an interval from 0 to
2(n-2), representing the number of edges in the topology, thus 38 was
attributed to identical trees, and 0 for trees sharing no bipartition
-since the rationale of this distance is to deem
every tree branch
as a bipartition to all leaves-. The resultant topologies were
different between each other, taking values from 10 to 36.
Relatively, the pairs of COI and MDH topologies had the lowest values
of this measure. As shown in the previous section of nodes recovery,
the R&F distance did not variate among the three weighting
schemes.
 |
|
Fig
3. Values of Bootstrap score for the topologies build from
different weighting schemes , based on the sum of the bootstrap
values of all of the nodes of the topologies. The implied
weighting scheme (IP) yielded the highest scores for the genes IDH
and MDH.
|
Bootstrap
support
Considering
that all nodes should contribute equally to the topology, I
calculated a bootstrap score, as the ratio of the sum of the support
of all nodes and the number of nodes of the topology (See
supplementary material). I distinguished the attribute of topologies
as follows: 100 as the maximal tree support, 90-99 as strong support,
65–89 as moderate support, 50–64 as weak support, and <50 as
negligible tree support (Lu et al., 2013). The topologies based on
nuclear genes essentially resulted in moderate values of support,
while the COI-based topologies showed weak values of support.
Regarding the weighting scheme, the results did not show differences.
Limitations
and Flaws
The
experiment per se had several failures. Since the purpose of
the study was to assess the influence of the weighting scheme over
the phylogenetic reconstruction, the first step would have been using
a different reference topology that could somehow group the different
partitions into one sequence to thence consider it as the “complete”
evidence. On the other hand, the range of concavity constant was
definitely an eye pad. This was reflected in the little differences
between each scheme. As recommended by other authors, it is
reasonable to explore different values of k, to thus compare whether
the resultant taxonomic groups are indeed well established or not
(Goloboff et al., 2008). Although I decided to count gaps as
characters, I did not consider to assign specific costs to the codon
positions, which have been proved to affect the final results (Rota,
2011). Finally, the bootstrap score calculation was perhaps a good
approach to assess support as a full-topology attribute.
Nevertheless, I did not applied a significance test on the data,
thence it was not sane to make any asseveration from those results,
but the assignation of attributes for instance, strong or moderately
supported.
Implications
Despite
all of the aforementioned, I found some interesting results. First,
the COI
gene showed that it
is
not suitable to reconstruct the
phylogeny of Pieridae under the weighting schemes I used. This,
probably because it is a highly conservative gene (Lunt et al.,
1996), which explains why it does not recover a correct phylogeny.
This result is consistent with other studies that evaluate the
phylogeny of a group, using different genome partitions (
Heikkilä
et al., 2011; Chen
et al., 2013). Second,
unlike
this gene, the EF-1a
showed a different behavior in the different reconstructions, which
is also consistent with
Heikkilä
et al., 2011.
For each method, it managed to recover all the tribes from the study
of Wahlberg et al., 2014 (See supplementary Material. Fig 1.).
Accordingly,
using this partition in further studies might be useful to yield
reasonable results with a small part of the genome. As regards the
weighting scheme, molecular data should be carefully managed to
consider every partition as a single different unit for the analysis,
thence a prior exploration of k values is convenient. Notwithstanding
the poor resolution of the results presented here, it has been proved
for other groups, that the weights of partitions do affect the
support and stability of the phylogenetic reconstructions (Chen et
al., 2013).
Supplementary Material
Fig 1S. Resultant topologies for each partition.

References
Allard,
M. W., & Carpenter, J. M. (1996). On weighting and congruence.
Cladistics, 12, 183–198.
Arias,
J.S., Miranda-Esquivel, D.R. (2005) "Yuu-PRC", macros para
TNT distributed by authors. Universidad Industrial de Santander,
Bucaramanga (Colombia).
Braby,
M. F., Vila, R., & Pierce, N. E. (2006). Molecular phylogeny and
systematics of the Pieridae (Lepidoptera: Papilionoidea): Higher
classification and biogeography. Zoological Journal of the Linnean
Society, 147(1986), 239–275. doi:10.1111/j.1096-3642.2006.00218.x
Carpenter,
J. M. (1994). Successive eighting, reliability and evidence.
Cladistics, 10, 215–220.
Edgar,
R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy
and high throughput. Nucleic Acids Research, 32(5), 1792–1797.
doi:10.1093/nar/gkh340
Farris,
J. S. (1969). A Successive Approximations Approach to Character
Weighting. Syst Biol, 18(4), 374–385.
Goloboff,
P. (1998). Tree Searches Under Sankoff Parsimony. Cladistics, 14(3),
229–237. doi:10.1006/clad.1998.0068
Goloboff,
P. a. (2014). Hide and vanish: Data sets where the most parsimonious
tree is known but hard to find, and their implications for tree
search methods. Molecular Phylogenetics and Evolution, 79, 118–31.
doi:10.1016/j.ympev.2014.06.008
Goloboff,
P. A. (1993). Estimating character weights during tree search.
Cladistics, 9, 83–91.
Goloboff,
P. A. (1997). Self-Weighted Optimization: Tree Searches and Character
State Reconstructions under Implied Transformation Costs. Cladistics,
13(3), 225–245. doi:10.1111/j.1096-0031.1997.tb00317.x
Goloboff,
P. A. (2013). Cladistics Extended implied weighting, 1–13.
Goloboff,
P. a., Carpenter, J. M., Arias, J. S., & Esquivel, D. R. M.
(2008). Weighting against homoplasy improves phylogenetic analysis of
morphological data sets. Cladistics, 24(5), 758–773.
doi:10.1111/j.1096-0031.2008.00209.x
Goloboff,
P. A., Farris, J. S., & Nixon, K. C. (2008). TNT, a free program
for phylogenetic analysis. Cladistics, 24(5), 774–786.
doi:10.1111/j.1096-0031.2008.00217.x
Heikkila,
M., Kaila, L., Mutanen, M., Pena, C., & Wahlberg, N. (2012).
Cretaceous origin and repeated tertiary diversification of the
redefined butterflies. Proceedings of the Royal Society B: Biological
Sciences, 279(September 2011), 1093–1099.
doi:10.1098/rspb.2011.1430
Hennig,
W. (1968). Elementos de una sistemática filogenética. Buenos Aires:
Eudeba.
Neff,
N. a. (1986). A Rational Basis for a Priori Character Weighting.
Systematic Biology, 35(1), 110–123. doi:10.1093/sysbio/35.1.110
Nixon,
K. C., & Carpenter, J. M. (2011). Cladistics On homology, 27,
1–10.
Nixon,
K. C., & Carpenter, J. M. (2012). Cladistics On homology, 28,
160–169.
Patterson,
C. (1988). Homology in Classical and Molecular Biology. Molecular
Biology and Evolution, 5(6), 603–625.
Rodrigo,
A. G. (1989). An information-rich character weighting procedure
for parsimony analysis. New Zealand Natural Sciences: 16-97, 103.
Sneath
R.R, S. P. H. A. (1973). Numerical Taxonomy. Freeman, San Francisco.
Swofford,
D. L., & Maddison, W. P. (1987). Reconstructing ancestral
character states under Wagner parsimony. Mathematical Biosciences,
87(2), 199–229. doi:http://dx.doi.org/10.1016/0025-5564(87)90074-5
Wahlberg,
N., Rota, J., Braby, M. F., Pierce, N. E., & Wheat, C. W. (2014).
Revised systematics and higher classification of pierid butterflies
(Lepidoptera: Pieridae) based on molecular data. Zoologica Scripta,
1–10. doi:10.1111/zsc.12075